
萧箫整理自 MEET
2023 量子位公众号 QbitAI
AI 制药,一个被称作明星赛道的行业。
不仅融资消息隔三差五传出,更被认为是计算生物最有希望落地的领域。
据量子位智库预测,AI 制药行业国内市场保守估计将达到 2040 亿元规模。
在这样的前景下,不仅国内外高校博士生和教授纷纷投身创业,就连互联网大厂们也争相入资角逐。
如今 AI 制药步入热度高峰后的瓶颈突破期,各玩家的差异性也逐步显现。
作为国内互联网巨头的腾讯,相比间接投资,三年前选择直接成立 AI 制药平台,成为赛道上角逐玩家之一。
现在,腾讯 AI 制药平台成果进展如何?相比同赛道玩家,其竞争优势是否得以体现?
在 MEET 2023 大会上,腾讯医疗健康 AIDD 技术负责人刘伟,从腾讯制药 AI 算法实践的角度探讨了这一行业当前的现状。

为了完整体现刘伟的分享及思考,在不改变原意的基础上,量子位对他的演讲内容进行了编辑整理。
关于 MEET 智能未来大会:MEET 大会是由量子位主办的智能科技领域顶级商业峰会,致力于探讨前沿科技技术的落地与行业应用。今年共有数十家主流媒体及直播平台报道直播了 MEET2023 大会,吸引了超过 300 万行业用户线上参会,全网总曝光量累积超过 2000 万。
演讲要点
- 在 AlphaFold 和 AlphaFold2 驱动下,AI 药物行业发展速度非常快,而腾讯在蛋白质结构预测上的工作从 2019 年初就开始了。
- 骨架跃迁分子生成算法能在保证原有化合物活性的基础上,突破原有分子专利的保护,或者优化分子的 ADMET 性质。
- 只靠数据驱动的 AI 缺乏可解释性,要与领域知识相结合;药物 AI 是 AI 算法和领域知识相互发现,相互提升的过程。
- AI 辅助药物发现存在一个难题就是泛化性,即在A靶点中训练的 AI 模型,通常难以应用到B靶点上做预测。这个问题的解决对技术的突破至关重要。
(以下为刘伟演讲分享全文)
腾讯云深智药是一个怎样的平台?
我分享的题目为“腾讯制药 AI 算法实践”,会对腾讯过去 3 年积累的 AI 制药技术进行一个展示。
我叫刘伟,是腾讯 AI 药物发现这一块的技术负责人。今天我要讲的内容,主要包括三个部分:
第一部分是腾讯云深平台介绍,它实际上是沉淀了腾讯 AI 制药技术的一个平台;第二部分是平台的案例分享;第三部分总结平台的技术优势。
我们先介绍一下腾讯云深平台的两大功能模块,一个是小分子药物发现,另一个是大分子药物发现,这里主要指抗体药物发现。
其中,小分子模块包括蛋白质结构预测、分子生成等 6 个模块,大分子模块则由抗体结构预测、抗体亲和力、以及抗体人源化改造等几大模块组成。

时间有限,我们不会详细介绍每一个模块,主要会讲案例以及部分底层技术的实现。
腾讯云深平台四大案例分享
第一个案例是蛋白质结构预测,这块腾讯在国内可能是做得最早的。
最近在 AlphaFold 和 AlphaFold2 的驱动下,行业发展速度非常快,而腾讯从 2019 年初就开始做相关技术工作。
大家应该比较清楚,蛋白质是生命及其活动中非常重要的组成部分,蛋白质结构预测则是指给定一个氨基酸序列,来预测蛋白质的三维结构。
我们在 2019 年打造的一个预测 pipeline,当时是基于分子动力学模拟和 AI 预测相结合的方法,构建了一个叫 tFold 的蛋白质结构预测平台。
在 2020 年比赛的时候,这个 pipeline 连续八周在评测平台排名第一。
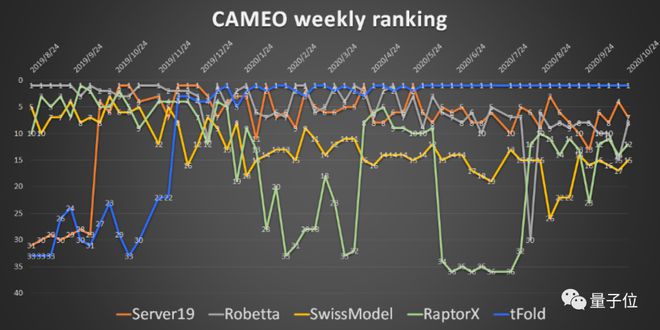
随后我们还参加了 CASP 竞赛,在国内获得了第一名,颜宁教授在她的论文中还引用了 tFold 平台,这是对我们在蛋白质结构预测上的成果的重要肯定。
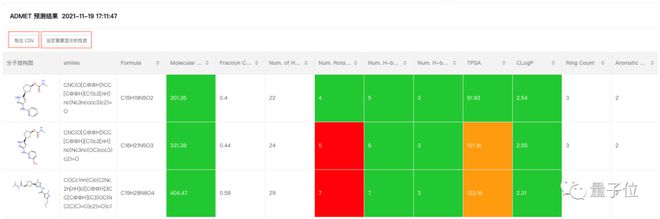
第二个案例是结合物理学特征和本地数据训练,我们做了ADMET 性质预测系列模型。
也是从 2019 年开始的工作,想跟大家分享两点:第一点是效果提升,以水溶性模块为例,我们做的 ADMET 相比头部商业软件效果平均会有 20% 的提升;第二点是 ADMET 预测成熟度,在与某药企合作上,我们会提供一个可以说是非常基础的模型,在药企拿到这样的模型之后,会基于具体项目的管线生成的很多内部数据,即项目和靶点相关的特定数据。
用这些数据对基础模型进行重新训练(retrain)、或者说做微调(finetune)后,它会有一个更好的提升。
比如我们与某个药企合作分析某系列化合物的心脏毒性,发现在项目中后期阶段,基本上与实验结果相关性达 95% 以上,后来药企就不太需要去做实验了,而是直接使用模型预测结果去做分析了。
所以我们在 ADMET 预测这块已经做到非常成熟,目前模型包含 60 多个属性预测模块,在腾讯云深平台上可以直接使用。
第三个案例是骨架跃迁分子生成算法,这也是针对国内药企或者国内科研机构的一些实际需求打造的,目前同样已经做得比较成熟。
通过骨架跃迁分子生成算法,就能在保证原有化合物活性的基础上,突破原有分子专利的保护,或者优化分子的 ADMET 性质。
我们自研了骨架跃迁算法,与药企做了一些合作,这些合作不仅帮助到药企发现了纳摩尔级别的多个化合物系列,也很好地验证了我们这个流程的健壮性,在不同的靶点、不同的复合物上都实现了比较好的效果,目前部分研究成果已经发表在期刊上。

第四个案例是将强化学习引入药物小分子的生成。
在生成分子的时候,我们不仅要考虑分子活性,还要考虑 ADMET 的属性,所以我们把这些流程做了一个打通,你可以定制这些 ADMET 属性的要求。
在生成过程当中,我们用上强化学习,使得生成出来的分子符合定制的属性要求,两个不同的模块能够相互提升和强化,最终建设一个非常完整的 pipeline,这可以用在各种分子生成的场景中。
例如这个例子中,生成有两个要求,包括不能通过血脑屏障、以及 logP 的属性:
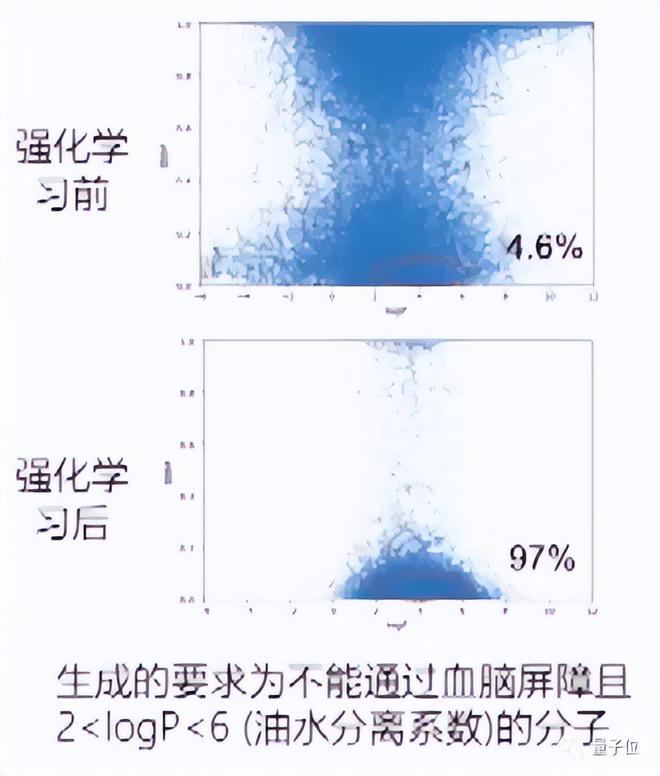
可以看到,在没有经过强化学习时,它的分布是比较弥散的,生成出来的化合物不太满足实际应用需求。但经过几轮强化迭代后,97% 的分子都会满足生成的要求。
这一平台具备哪些技术优势?
接下来,我们总结一下腾讯云深经过两三年发展后,积累出的一些技术优势。
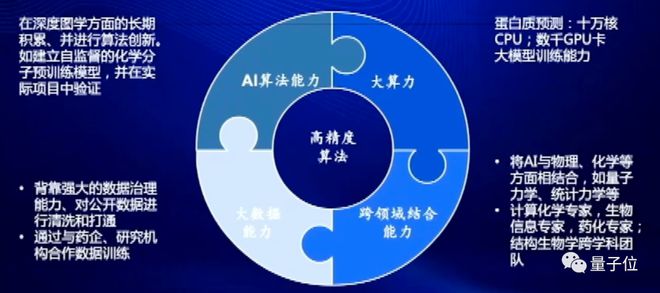
第一块是在AI 算法方面。大家知道,在药物研发这一块最主流的技术就是深度图神经网络,腾讯在做药物 AI 之前,在这方面做了非常长时间的研发,也有非常深厚的技术积累。
因为腾讯是一个社交网络公司,所以在深度图神经网络方面有深厚的研发积累,包括在一些 AI 顶会如 NeurIPS 上面,我们腾讯 AI Lab 在上面发表了非常多的论文,包括大规模图随机采样、以及图自监督学习等,其中一些论文的引用量非常高。
第二块和第三块,就是大算力、大数据方面的能力。
我们知道化学空间非常大,以 10 的n次方为计数,在这么大的化学空间中发现药物分子非常不容易。
通常的做法是借鉴自然语言处理或者图像视觉技术发展而来的大模型预训练,使得模型本身能够理解化学空间的语言,比如像理解 SMILES、3D 分子结构,甚至是理解蛋白质结构。
所以这方面会面临针对大模型、大算力的强劲需求,我们在这一块也做了非常多的工作,后面会详细介绍。
最后一块,我们基于腾讯在算法算力上的能力,将AI 与物理、化学做了一个结合,这是团队新生长出来的能力。
我们认为,AI 纯粹只靠数据驱动是不足够的、缺乏可解释性的,做出来模型之后,它有时也会因为数据稀疏、漂移产生一些问题。如果能够结合物理、化学知识进入 AI 算法模型,就能够非常好地反映在化学、或是底层物理方面的一些特征和规律。这样做出来的 AI 模型,不仅过拟合风险更低,实际应用中也有非常好的可解释性,这也是我们最近几年重点发展的一个能力。
我们再展开给大家分享一下这几块内容。
第一块是 AI+ 量子化学方面的算法能力。
我们开发了一系列用 AI 方法做量子化学计算的算法。量子化学是一个非常广泛而深刻的技术领域,它从比较低精度的经验性计算到非常高精度的多体计算,都已经开发出了很多方法。
对于现存最高准确度的,例如全组态量子化学计算,它需要非常大的计算量,这对像药物分子、或者说 drug-like 这样的分子是不太可行的,只能被迫使用比较低精度的方法。
针对这样的痛点,我们做了一个叫DeepQC的框架,可以在以秒为量级的时间单位下,达到高精度大基组 DFT 的计算结果。
我们还把 DeepQC 用在像晶体、催化体系上,也取得了非常好的效果,特别值得一提的是,在今年的催化剂相关比赛 Open Catalyst Challenge 2022 上我们还拿了冠军。
这也是基于我们多年以来对 AI 和量子化学结合的探索基础之上做出的工作。
这样一套方法,我们原本只是在有机分子上,我们看到将其推广到催化的表面体系,以及晶体的周期体系,它仍然可以非常高精度、非常快速地完成任务,它的速度比用量子化学模拟要快一个数量级。
第二块是我们在图卷积神经网络方向的技术发展。
腾讯以前做的很多图卷积神经网络工作在社交网络方面,但我们把它拓展到蛋白质结构上,发现也有非常好的效果。
实际上不管是蛋白质,还是这些配体和药物体系里面,它其实都有非常好的层次结构,和社交网络非常相似。
蛋白质通常被分为几层结构。从最底层的原子、甚至电子,再到氨基酸,氨基酸又组成蛋白质多肽链,再到上面三级结构或四级结构,如果在各种不同的层级用不同神经网络建模方法,就可以把它做得更好。

我们把层次图卷积神经网络用在抗体结构预测上,效果超过了 AF2、IgFold 等模型的结果,目前相关论文也已经被 NeurIPS 收录,也已经在 arXiv 上公开。
第三块是大分子预训练模型。
我们刚才提到,药物化学空间非常大,但是具体到某个药物研发项目中时,数据非常少甚至没有,所以你必须解决过拟合的问题。
我在做某一个项目、某一个靶点的时候,它的数据可能就只有几百甚至几十个,这种情况下如果没有大数据作为基础,是非常容易过拟合的。
我们在 2019 年就意识到这样的问题,发表了一个大规模分子预训练模型叫 GROVER,现在几乎这个领域的所有工作,都会引用当初我们在 GROVER 上做的成果。
我们是国内最早在分子图上做预训练的,而这也是腾讯云深平台非常底层的技术,不管是在分子属性预测、还是蛋白质结构预测、还是抗体药物设计上,它都是非常底层的公共技术模块。
不管是针对 2D 还是 3D 分子,都会基于 embedding 做特征提取,然后也是预训练+微调这样来用,也是行业内的一个范式。
最后,就是我们今年最新的一个工作。
我们在做 AI 辅助药物发现时,发现了这样一个问题,在A靶点(A场景)中训练的 AI 模型,会非常难以应用到B靶点(B场景)上做预测。
这个其实就是 OOD(out-of-distribution)问题。这个是机器学习自身的一个核心问题,现在也没能 100% 完全解决。
为了验证我们模型的有效性,我们去做了一个名叫 DrugOOD 的开源框架,现在它也已经贡献给了行业和社区。

我们会根据不同的 domain 把它做一个划分,比如按照骨架、实验 assay、或者靶点区分,这样训练出来的模型就会非常不一样。
这个过程中,我们实际上希望模型在不同场景具备一定的迁移能力,不然模型只能适合某一训练数据场景,这其实不是我们所希望的,因为这样的模型适应能力非常弱,没办法应用到新的问题上。
在 DrugOOD 中我们会有一个数据 Curator 的模块,之后我们会做一个分割,这样在训练不同模型时,我们就可以按照不同的标注去自动写一个配置文件,测定我们新训练的模型在不同的蛋白质家族上不同的效果,这样对模型的泛化性就能有一个非常明确的认识。






