
西风发自凹非寺
量子位公众号 QbitAI
把 169861 个生物物种数据装进大模型,大模型竟 get 到了生物中心法则的奥秘——
不仅能识别 DNA、RNA 与相应蛋白质之间的内在联系,在基因分类、蛋白质相互作用预测、热稳定性预测等 7 种不同类型任务中也能比肩 SOTA 模型。
模型名为 LucaOne,由阿里云飞天实验室生物智能计算团队打造。
相比 AlphaFold 3 因未开源遭到 650 多名学者联名批评,LucaOne 训练推理代码及相关数据目前均已开源。
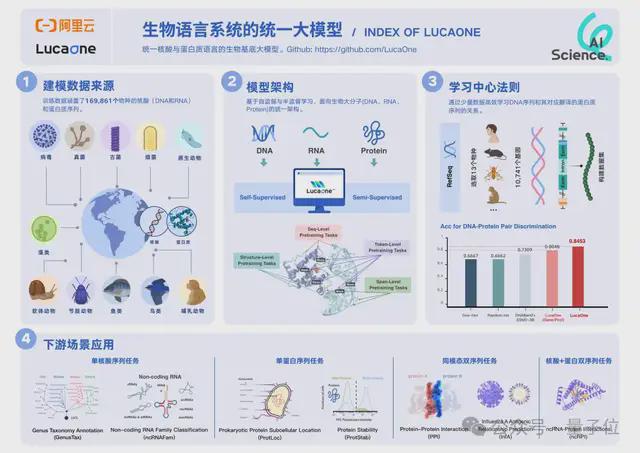
LucaOne 是目前首个全生物系统的核酸语言+蛋白语言的融合基座模型。换句话说,LucaOne 由核酸(DNA、RNA)和蛋白质序列联合训练而来。
通过一系列实验,研究人员发现它能广泛适用各种下游任务。
在含 13 个物种、关系对总数量为 24000 的核酸序列和其对应蛋白的正负样本数据集中,LucaOne 提供表征的模型达到 0.85 的预测准确率。
远高于目前业内最好的预训练模型组合 ESM-3B+DNAbert2(0.73)及其他建模方式,也显著高于 LucaOne 的单核酸训练版本+单蛋白训练版本。
其他任务如针对流感 H3N2 病毒疫苗有效性(免疫逃逸风险)的预测,LucaOne 准确率可达 100%。

量子位也联系到了论文一作,聊了聊 LucaOne 的实现细节,以及 AI for Science 在生物科学领域的发展。

核酸和蛋白质序列联合训练
总的来说,LucaOne 围绕中心法则的数据进行构建,能够学习到中心法则背后的原理和逻辑,可提取基因转录和蛋白质翻译过程中固有的复杂模式和关系,在使用层面相当于提供了一个对 DNA、 RNA、蛋白质的无差别表征。
PS:
分子生物学的中心法则即遗传信息从 DNA 传递给 RNA,再从 RNA 传递给蛋白质的过程,这一过程包括 DNA 的复制、RNA 的转录和蛋白质的翻译。
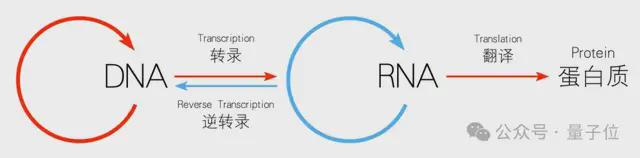
展开来看,LucaOne 整个工作流是这样婶儿的:
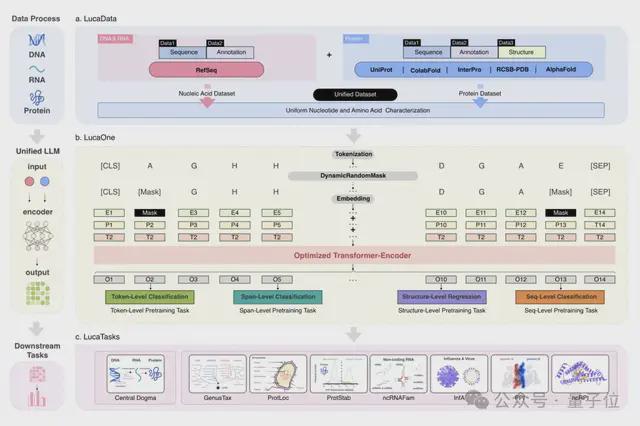
从技术上讲,构建 LucaOne 的难点首先是数据集的构建。
在生命科学领域,真实存在的只是分子数据。
例如,核酸的表示方式是 4 种碱基。DNA 是腺嘌呤(A)、鸟嘌呤(G)、胞嘧啶(C)和胸腺嘧啶(T);RNA 是腺嘌呤(A)、鸟嘌呤(G)、胞嘧啶(C)、尿嘧啶(U)。蛋白质由氨基酸组成,自然界存在的氨基酸大约有 20-22 种,每种氨基酸也用一个字母表示。
而人类为理解这些分子的性质与作用,通常需要添加很多注释信息,包括一些图片的注释。注释信息属于人类语言,自然界本身不存在,从而就形成了一种生命科学领域的从“自然界”语言到“人类文化”语言的跨模态。
因此,LucaOne 的预训练数据不仅包含 DNA、RNA、蛋白质这三类分子的序列(核苷酸序列或者氨基酸序列)数据,同时还使用了这些分子的注释信息。
总共涵盖了 169861 个物种的核酸和蛋白质序列和注释信息,分为两部分:
核酸数据集来自 RefSeq,包括核酸序列及注释;蛋白质数据集来自 InterPro、UniProt、ColabFold、RCSB-PDB、AlphaFold2,包括蛋白质序列、注释和三维结构。
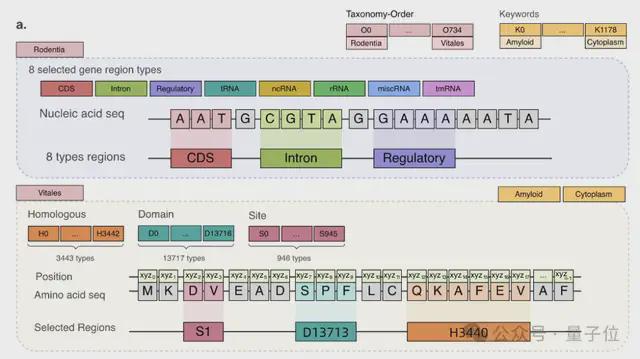
据介绍,在数据集的收集处理方面,阿里云飞天实验室与中山大学、浙江大学等多个团队进行了合作。
另一大难点是生物分子序列的预测和大语言模型预测下一个 token 不同,在模型训练阶段还需要一些专门的设计。
LucaOne 采用了 Transformer-Encoder 架构,由 20 个编码器块组成,嵌入维度为 2560,总参数量 1. 8B。
研究人员在此基础上进行了一些优化:
- 使用 Pre-Layer 归一化代替 Post-Layer 归一化,以便更好地训练深层网络;
- 使用旋转位置嵌入(RoPE)代替传统绝对位置编码,以推理更长序列。
此外,在数据处理和模型训练过程中,核苷酸和氨基酸用统一的方式进行表征或编码。通过 token-type embeddings 实现核酸和蛋白质序列的混合训练,区分核苷酸(0)和氨基酸(1)。
在两个自监督掩码任务的基础上,研究人员还增加了八个半监督预训练任务,通过序列注释增强模型对数据的理解。
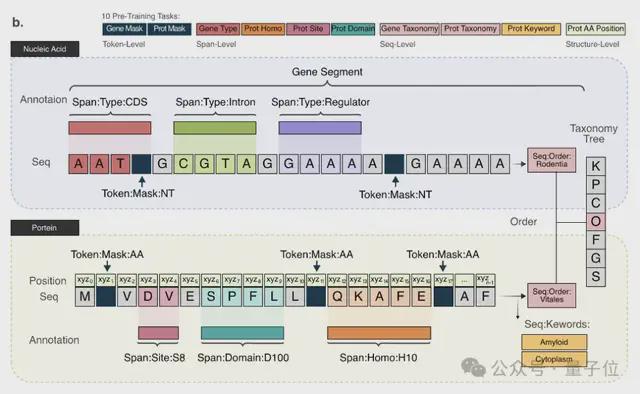
已能理解基因和蛋白对应关系
为验证核酸和蛋白质数据混合训练的优势,研究人员分别使用核酸和蛋白质数据单独训练了两个额外的模型——LucaOne-Gene 和 LucaOne-Prot,并使用相同的 5.6M checkpoint 在分子生物学中心法则任务中进行了比较。
使用t-SNE 可视化说明,与其他模型相比,LucaOne 的嵌入在两个数据集上呈现出更紧密的聚类,可能包含了更多上下文信息。
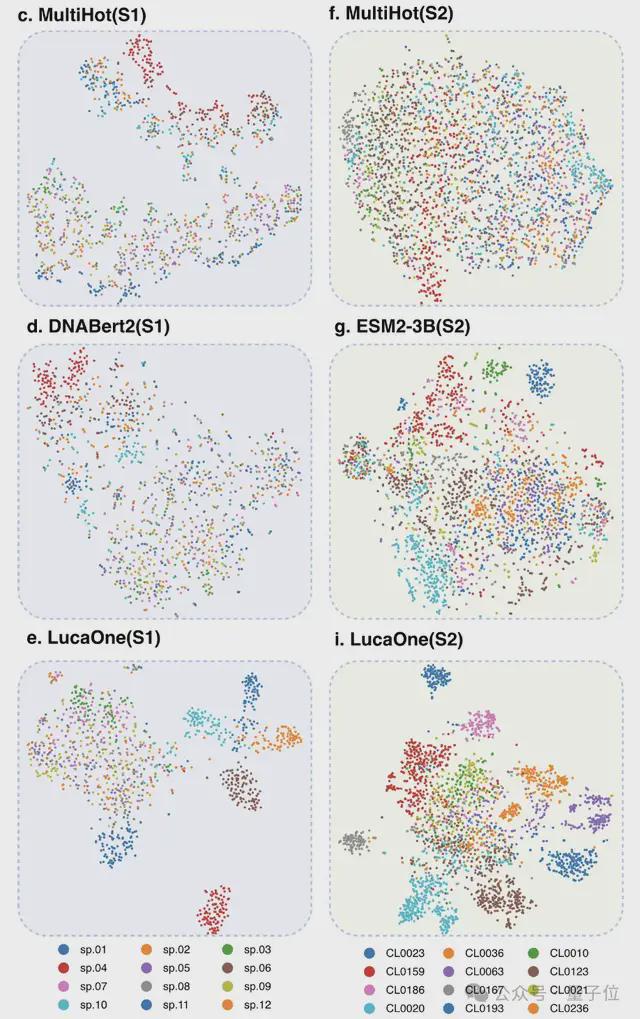
为验证 LucaOne 通过广泛学习基因及蛋白语言,已具备对生物学中心法则里的基因和蛋白对应关系的理解能力,研究人员设计了一个数据集及评测任务。
选取 13 个物种的核酸序列和其对应蛋白的正负样本数据集,关系对总数量为 24000,其中正负样本比例1:2。基因序列数据是其在基因组的原始数据,包括了大量的非编码区(内含子,调控元件,及“垃圾片段”等)。
采用训练:验证:测试比例为:4:3:25;即仅 3200 组数据作为训练,18750 组数据作为测试集来预测其核酸序列是否可以翻译成数据组里的蛋白序列。
结果 LucaOne 提供表征的模型达到 0. 85 的预测准确率,不仅远高于目前业内最好的预训练模型组合 ESM-3B+DNAbert2(0.73)及其他建模方式,也明显高于 LucaOne 的单核酸训练版本+单蛋白训练版本。
这表明这两种大分子数据联合训练可以显著增强模型的学习效果。
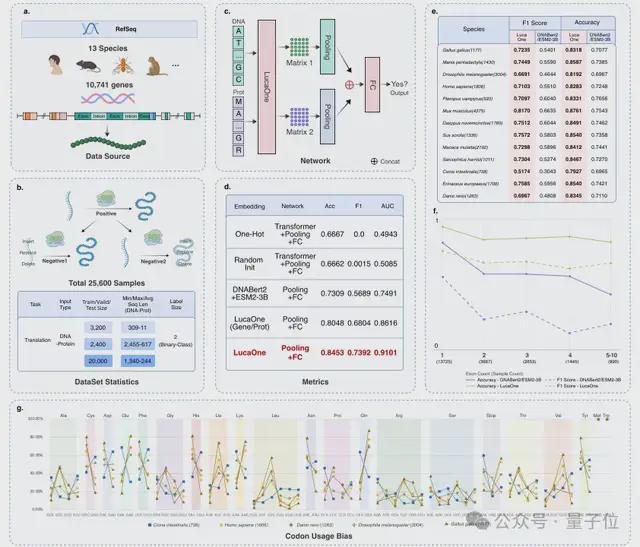
有意思的是,研究人员发现在模型细分表现里,LucaOne 海鞘这种生物里的预测表现比较差(其他模型也类似),进一步分析海鞘的特性表明,因为进化适应性等各种原因,海鞘利用中心法则的具体规则-密码子偏好性,和其他生物明显不同。
他们猜测 LucaOne 可能用的是另一种中心法则语法“方言”,而这种“方言”在训练数据集里仅有 100 条,因此模型没有很好的学习到这种规则。
在其他下游任务中,LucaOne 对不同类型输入的下游任务也广泛适用。
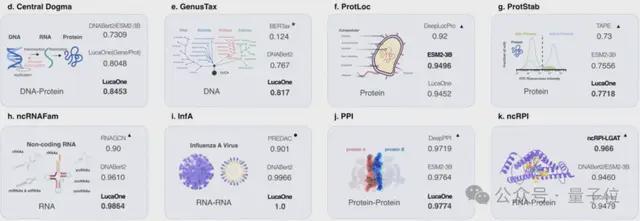
具体来说,研究人员评估了 7 个不同类型的下游生物计算任务,包括:
- 单序列任务:GenusTax(属分类)、ncRNAFam(ncRNA 家族分类)、ProtLoc(蛋白质亚细胞定位)、ProtStab(蛋白质热稳定性预测)。
- 同源序列对任务:InfA(流感血凝素分析)、PPI(蛋白质相互作用预测)。
- 异源序列对任务:ncRPI(ncRNA-蛋白质相互作用预测)。
为简化下游任务,研究人员使用了三种对应不同输入形式的简单网络架构:
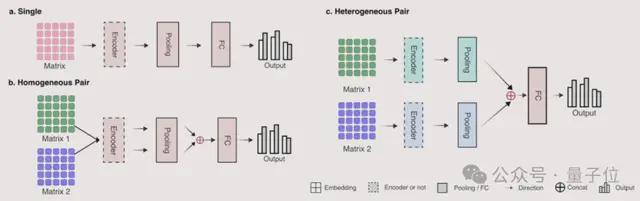
结果表明,GenusTax、ProtStab、ncRNAFam、InfA、PPI 任务上,LucaOne 显著优于其他模型;ProtLoc 任务上,LucaOne 与 ESM2-3B 相当,优于 SOTA;ncRPI 任务上,LucaOne 优于 DNABert2+ESM2-3B 的组合:
值得一提的是,在流感 H3N2 病毒的免疫逃逸风险预测中,研究人员采用了 1968 年至 2010 年间分离的大规模 H3N2 病毒 HA 序列数据进行了基于流感毒株抗原关系的预测模型。
通过病毒 HA 抗原序列来预测其是否会诱导 HIA 实验的血凝现象,进而预测其是否在特定人群中会发生免疫逃逸。
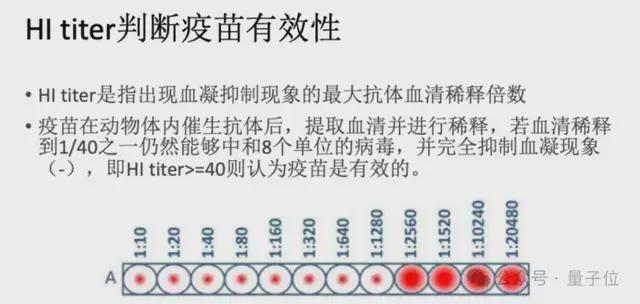
要知道,目前预防与控制流感最有效的方法是接种流感疫苗,但是由于流感病毒极快的变异速度,导致不能及时与准确地推荐与流行病毒相匹配的流感疫苗株。根据 WHO 和 CDC 的监测,流感疫苗的有效性在 40%-60% 之间。因此准确预测流行毒株,判断免疫逃逸风险是一个重要且困难的命题。
研究人员使用基于 LucaOne+ 一层感知机的模型达到了 100% 的准确率。
这也说明 LucaOne 学习的大量核酸序列信息,包括大量其他病毒序列,为计算特定任务提供了很好的信息补充。
更多细节,感兴趣的家人们可查看原论文。
“Science for AI”
如前文所述,LucaOne 背后开发团队来自阿里云飞天实验室 LucaTeam,LucaTeam 也与多个团队展开了深度合作。
中山大学医学院施莽教授及其团队参与了 LucaOne 模型的数据设计与验证。施莽教授认为:
LucaOne 是一项极为重要的尝试。最让我惊讶的是,在没有任何先验知识的前提下,LucaOne 确实能够更有效地学习中心法则中核酸与蛋白质之间的对应关系。
中国医学科学院北京协和医学院病原生物学研究所所长、美国微生物科学院会士舒跃龙教授及其团队参与了 LucaOne 在流感病毒方面的分析与验证工作。舒跃龙教授表示:
将前沿的 AI 技术与病原生物学相结合具有重大的科学意义和社会价值。通过这种紧密的跨学科协作,我们能探索更多病原生物起源进化、跨种传播以及感染致病等方面的规律,为传染病防控和生物安全做出更大的贡献。
此外,论文一作贺勇是阿里云飞天实验室生物计算高级算法专家,我们也就 LucaOne 与其展开聊了聊 AI for Science 在生物科学领域的发展。
在他看来,AI for Science 在生物科学领域正处于刚起步的阶段,考虑到 AI 可解释性的问题,基本上现在他们还只是把 AI 当做一个工具,但同时现在是发展 AI for Science 的一个很好的时间节点。
因为现在测序技术发展非常迅速,测序成本下降很多,分子序列数据很容易就能获得。有了数据大量的积累,就可以数据驱动的 AI for Science 相关工作。
不过,目前的局限在于测序得到还是分子的序列数据,而真实世界每个分子的存在是一个空间结构,这可能就需要更复杂的模型来处理。
而不同学科间的研究方法不同、探索微观世界也受限于目前的设备技术,贺勇认为人类对生物科学这个领域的认识目前也只是冰山一角,还无法从全局角度构建一个全面通用的系统。
最后他还补充道:目前大家对 AI for Science 越来越重视,相当于用 AI 去解决具体的问题。我想接下来还应该回过头来看 Science 能为 AI 带来什么?解决具体问题是第一步,最后通过问题反哺技术本身其实可能也是应该考虑的。
论文链接:https://www.biorxiv.org/content/10.1101/2024.05.10.592927v1
GitHub 链接:https://github.com/LucaOne






